Skip Navigation
Black Diamond Therapeutics
About
Our Vision
At a Glance
Executive Team
Board of Directors
Pipeline
Pipeline
Our Approach
BDTX-1535 NSCLC
BDTX-1535 GBM
BDTX-4933
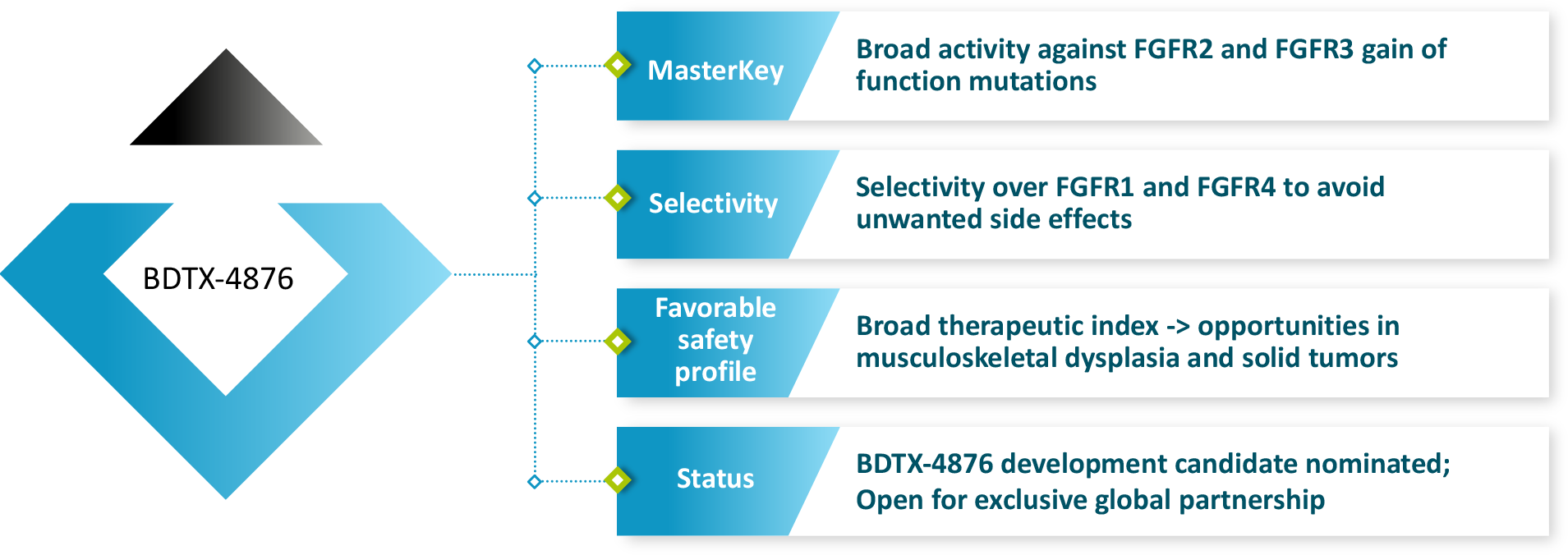
BDTX-4876
Presentations and Publications
Patients
Clinical Trials
Expanded Access
Investors & News
Overview
Press Releases
Events & Presentations
Corporate Governance
Stock Information
SEC Filings
Analyst Coverage
Investor Resources
Join Us
Values
Careers
BDTX-4876
You are about to leave the Black Diamond website.
Do you wish to continue?